Efficacy Data for WELIREG® (belzutifan) in Adults With Certain VHL Disease–Associated Tumors

Reimagine your approach to the management of certain VHL disease–associated tumors
When surgery is not immediately required, consider the only approved systemic option1,2


Nearly half of patients achieved tumor reduction in VHL disease–associated RCC with WELIREGa
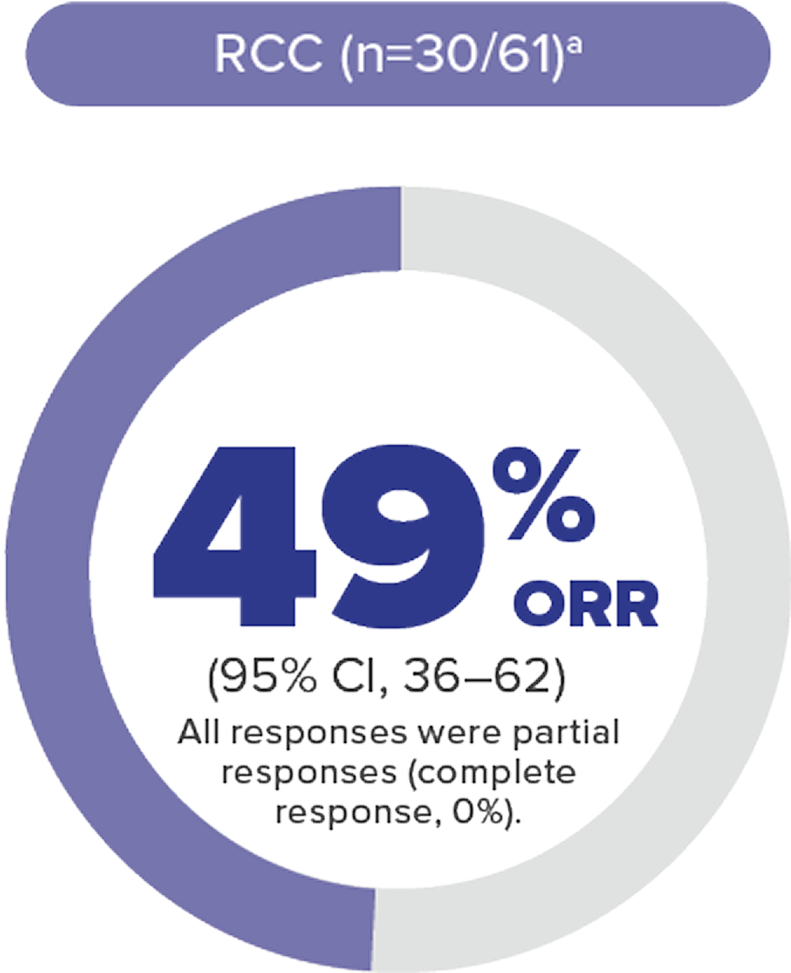
Objective response rate (ORR) per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Any pathological lymph nodes (whether target or nontarget) must have reduction in short axis to <10 mm. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4
MEDIAN DURATION OF RESPONSE WAS NOT REACHED
- Median DOR could not be estimated (by Kaplan-Meier method), as the majority of patients who responded to treatment maintained their response (did not experience disease progression per RECIST v1.1) at the time of data cutoff.5
56 %
- Median time to response was 8 months (range: 2.7 to 19 months).
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Kidney Cancer (confirmed hereditary VHL disease–associated RCC)
PATIENT SUBGROUP: VHL disease–associated CNS hemangioblastomas
Nearly two-thirds of patients with VHL disease–associated CNS hemangioblastomas achieved tumor reduction with WELIREGa
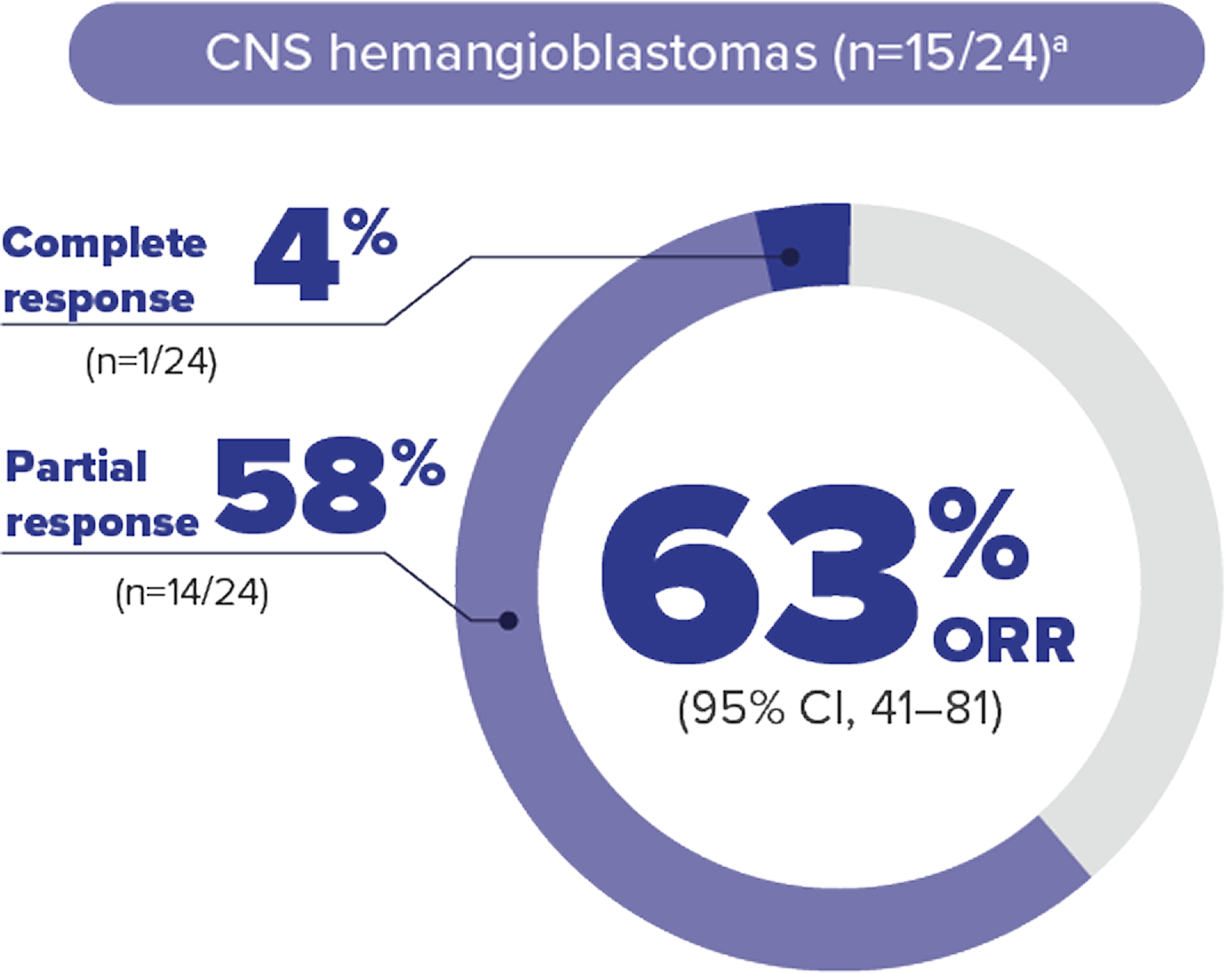
ORR per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Any pathological lymph nodes (whether target or nontarget) must have reduction in short axis to <10 mm. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4
MEDIAN DURATION OF RESPONSE WAS NOT REACHED
- Median DOR could not be estimated (by Kaplan-Meier method), as the majority of patients who responded to treatment maintained their response (did not experience disease progression per RECIST v1.1) at the time of data cutoff.5
73 %
- Median time to response was 3.1 months (range: 2.5 to 11 months).
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Central Nervous System Cancers (VHL disease–associated CNS hemangioblastomas)
PATIENT SUBGROUP: VHL disease–associated pNET
Most patients with VHL disease–associated pNET achieved tumor reduction with WELIREGa
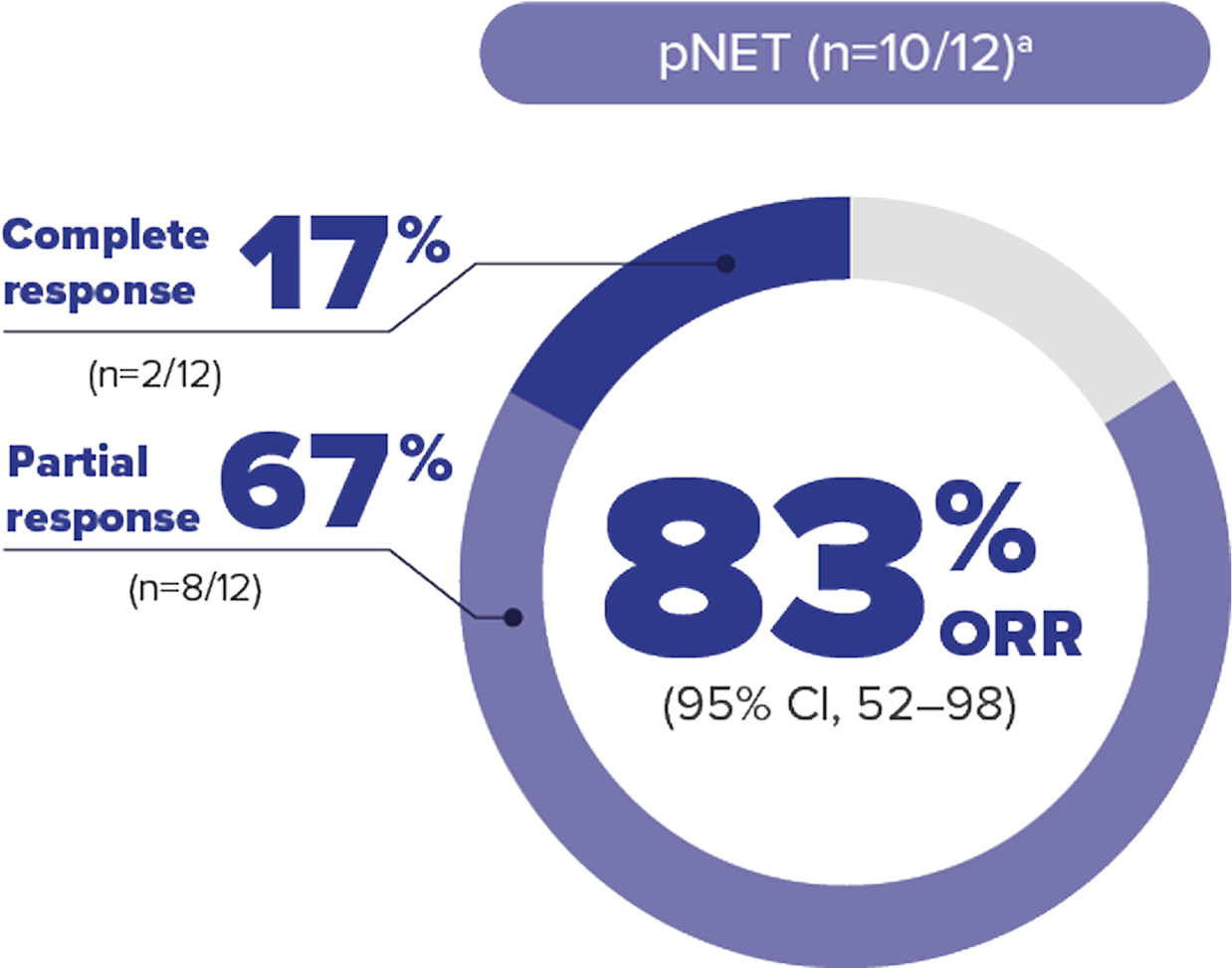
ORR per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Any pathological lymph nodes (whether target or nontarget) must have reduction in short axis to <10 mm. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4
MEDIAN DURATION OF RESPONSE WAS NOT REACHED
- Median DOR could not be estimated (by Kaplan-Meier method), as the majority of patients who responded to treatment maintained their response (did not experience disease progression per RECIST v1.1) at the time of data cutoff.5
50 %
- Median time to response was 8.1 months (range: 2.7 to 11 months).


Objective response rate (ORR) data at 61.8-month median follow up in adult patients with VHL disease–associated RCC8
LIMITATIONS: Approval of WELIREG is not based on the extended follow-up analysis; therefore, these data are for informational purposes only.
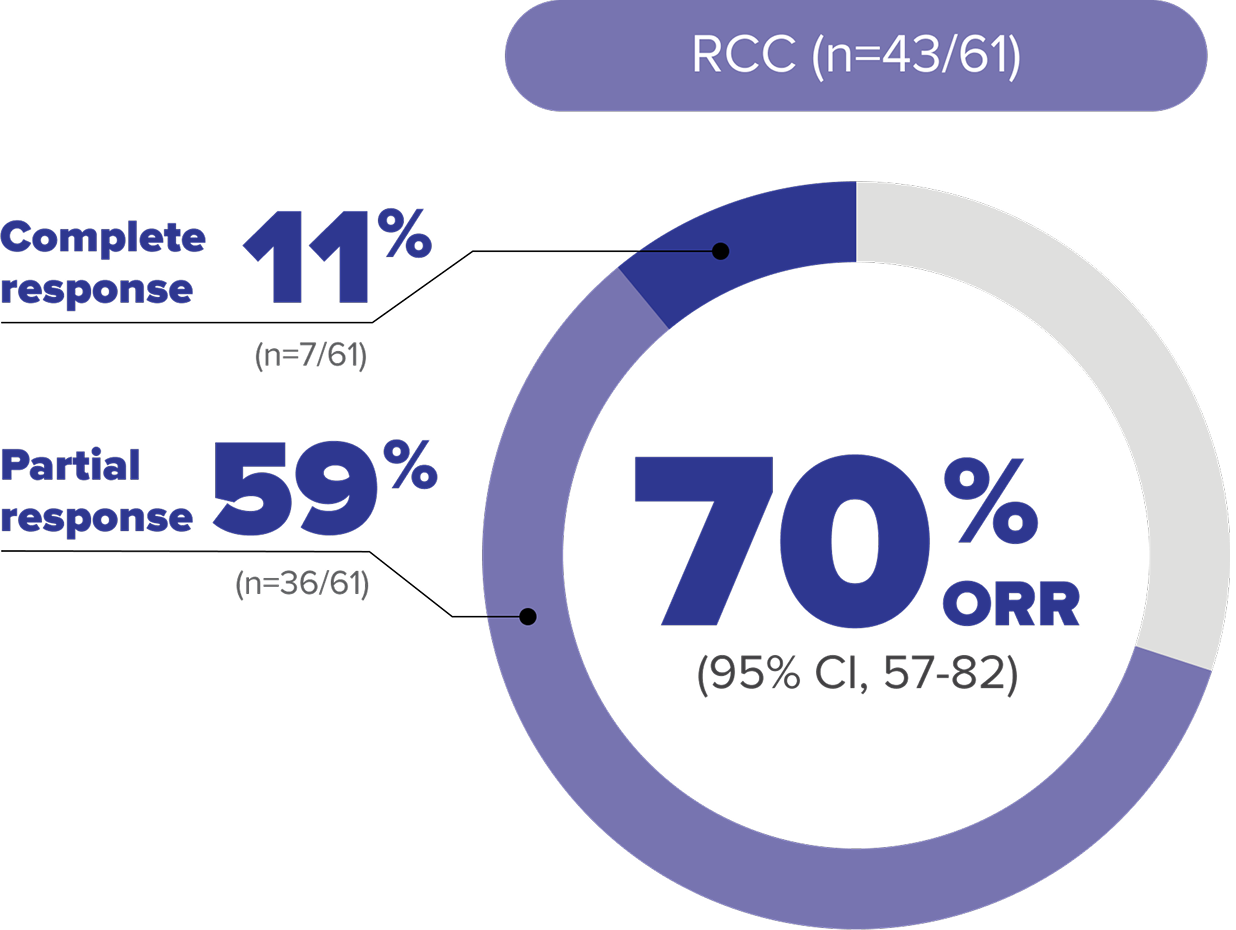
ORR per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4,8
MEDIAN DURATION OF RESPONSE WAS NOT REACHED
- Median DOR could not be estimated (by Kaplan-Meier method), as the majority of patients who responded to treatment maintained their response (did not experience disease progression per RECIST v1.1) at the time of data cutoff.5,8
Objective response rate (ORR) data at 61.8-month median follow up in adult patients with VHL disease–associated CNS hemangioblastomas8
LIMITATIONS: Approval of WELIREG is not based on the extended follow-up analysis; therefore, these data are for informational purposes only.
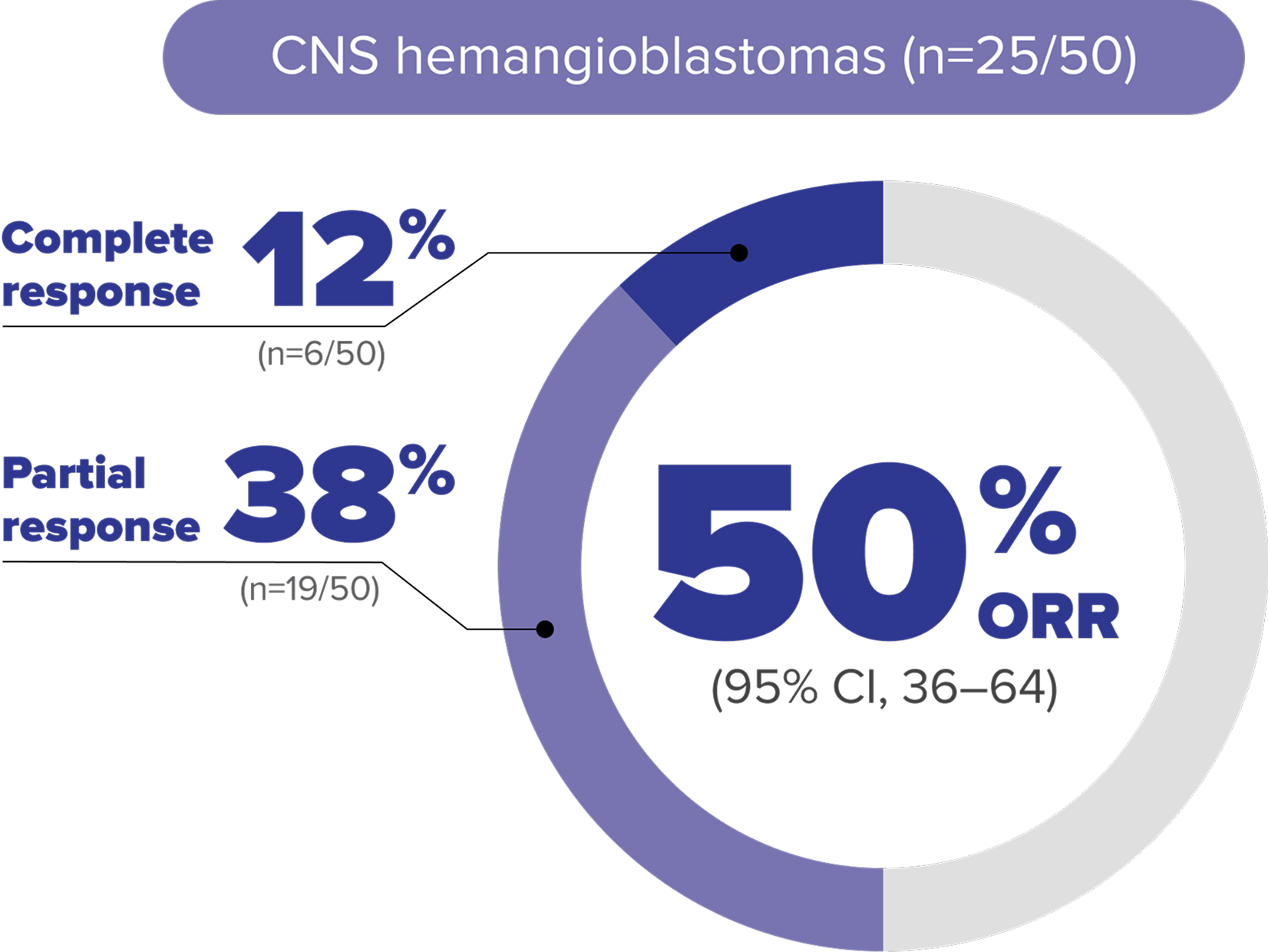
ORR per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4,8
MEDIAN DURATION OF RESPONSE WAS 60.3 MONTHS
In the extended analysis, the number of patients with CNS hemangioblastomas is from a prespecified data set that included solid lesions and the associated cystic component, if present. Other post hoc data that specifically assessed only solid lesions are not included here.10
Objective response rate (ORR) data at 61.8-month median follow up in adult patients with VHL disease–associated pNET8
LIMITATIONS: Approval of WELIREG is not based on the extended follow-up analysis; therefore, these data are for informational purposes only.
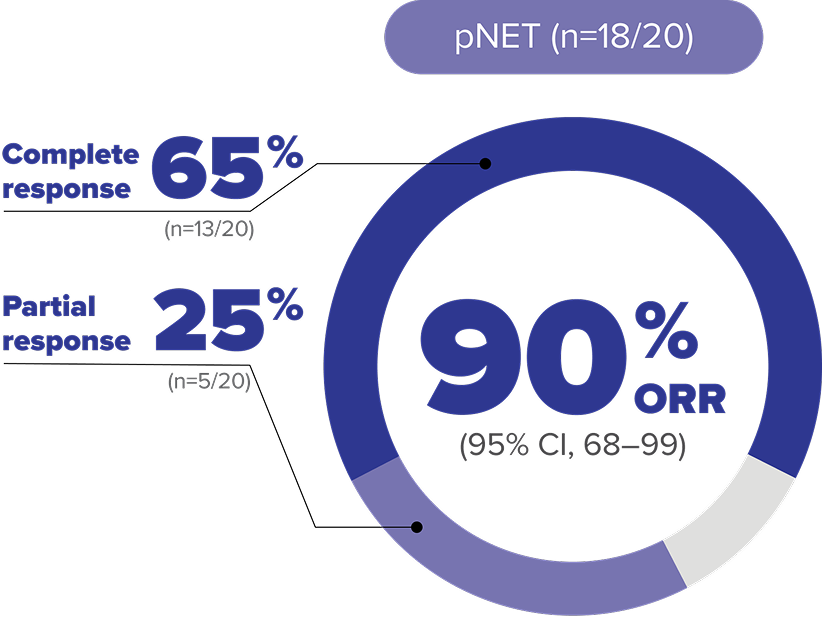
ORR per RECIST v1.1: Complete response was defined as disappearance of all target and nontarget lesions. Partial response was defined as ≥30% decrease in the sum of the longest diameters of target lesions compared with baseline.4,8
MEDIAN DURATION OF RESPONSE WAS NOT REACHED
- Median DOR could not be estimated (by Kaplan-Meier method), as the majority of patients who responded to treatment maintained their response (did not experience disease progression per RECIST v1.1) at the time of data cutoff.5,8
In the extended analysis, pNET was assessed by 2 different review committee approaches. The data set shown is by an IRC approach consisting of scan review by 2 primary blinded, independent radiologists, with a third radiologist as an adjudicator when needed. Other post hoc data that required the presence of pNET were confirmed by at least 2 of the 3 blinded radiologists' reviews and are not included here.11
Study design
WELIREG was evaluated in LITESPARK-004, an open-label clinical trial


Prior to enrollment in LITESPARK-004, 77% of study participants had previous surgical procedures for RCC
As defined by RECIST v1.1.
Assessed by IRC using RECIST v1.1.
CNS hemangioblastomas and pNET in these patients were diagnosed based on the presence of at least 1 measurable solid tumor in the brain/spine or pancreas, respectively, as defined by RECIST v1.1 and identified by IRC.
RECIST v1.1 response defined as ≥30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters.4
CNS = central nervous system; IRC = independent review committee; pNET = pancreatic neuroendocrine tumors; RCC = renal cell carcinoma; RECIST v1.1 = Response Evaluation Criteria In Solid Tumors v1.1; VHL = von Hippel-Lindau.
References: 1. Narayan V, Jonasch E. Systemic therapy development in von Hippel-Lindau disease: an outsized contribution from an orphan disease. Cancers (Basel). 2022;14(21):5313. 2. Center for Drug Evaluation and Research. Advancing Health Through Innovation: New Drug Therapy Approvals 2021. https://www.fda.gov/media/155227/download. Published January 2022. Accessed October 15, 2025. 3. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel–Lindau disease. N Engl J Med. 2021;385(22):2036–2046. 4. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. 5. Delgado A, Guddati AK. Clinical endpoints in oncology - a primer. Am J Cancer Res. 2021;11(4):1121–1131. 6. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Kidney Cancer V.1.2026. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed October 15, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. 7. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Central Nervous System Cancers V.2.2025. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed October 15, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. 8. Narayan V, Jonasch E, Iliopoulos O, et al. HIF-2α inhibitor belzutifan in von Hippel-Lindau disease–associated neoplasms: 5-year follow-up of the phase 2 LITESPARK-004 study. Poster presented at the 2025 American Society of Clinical Oncology (ASCO) Annual Meeting; May 30–June 3, 2025; Chicago, IL. 9. Narayan V, Jonasch E, Iliopoulos O, et al. Hypoxia-inducible factor-2α (HIF-2α) inhibitor belzutifan in von Hippel-Lindau (VHL) disease–associated neoplasms: 5-year follow-up of the phase 2 LITESPARK-004 study. In: Proceedings from the American Society of Clinical Oncology; May 30–June 3, 2025; Chicago, IL. Abstract 4507. 10. Iliopoulos O, Iversen AB, Narayan V, et al. Belzutifan for patients with von Hippel-Lindau disease-associated CNS haemangioblastomas (LITESPARK-004): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2024;25(10):1325–1336. 11. Else T, Jonasch E, Iliopoulos O, et al. Belzutifan for von Hippel-Lindau disease: pancreatic lesion population of the phase 2 LITESPARK-004 study. Clin Cancer Res. 2024;30(9):1750–1757.
References: 1. Narayan V, Jonasch E. Systemic therapy development in von Hippel-Lindau disease: an outsized contribution from an orphan disease. Cancers (Basel). 2022;14(21):5313. 2. Center for Drug Evaluation and Research. Advancing Health Through Innovation: New Drug Therapy Approvals 2021. https://www.fda.gov/media/155227/download. Published January 2022. Accessed October 15, 2025. 3. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel–Lindau disease. N Engl J Med. 2021;385(22):2036–2046. 4. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. 5. Delgado A, Guddati AK. Clinical endpoints in oncology - a primer. Am J Cancer Res. 2021;11(4):1121–1131. 6. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Kidney Cancer V.3.2025. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed June 12, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. 7. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Central Nervous System Cancers V.1.2025. © National Comprehensive Cancer Network, Inc. 2025. All rights reserved. Accessed June 6, 2025. To view the most recent and complete version of the guideline, go online to NCCN.org. 8. Narayan V, Jonasch E, Iliopoulos O, et al. HIF-2α inhibitor belzutifan in von Hippel-Lindau disease–associated neoplasms: 5-year follow-up of the phase 2 LITESPARK-004 study. Poster presented at the 2025 American Society of Clinical Oncology (ASCO) Annual Meeting; May 30–June 3, 2025; Chicago, IL. 9. Narayan V, Jonasch E, Iliopoulos O, et al. Hypoxia-inducible factor-2α (HIF-2α) inhibitor belzutifan in von Hippel-Lindau (VHL) disease–associated neoplasms: 5-year follow-up of the phase 2 LITESPARK-004 study. In: Proceedings from the American Society of Clinical Oncology; May 30–June 3, 2025; Chicago, IL. Abstract 4507. 10. Iliopoulos O, Iversen AB, Narayan V, et al. Belzutifan for patients with von Hippel-Lindau disease-associated CNS haemangioblastomas (LITESPARK-004): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2024;25(10):1325–1336. 11. Else T, Jonasch E, Iliopoulos O, et al. Belzutifan for von Hippel-Lindau disease: pancreatic lesion population of the phase 2 LITESPARK-004 study. Clin Cancer Res. 2024;30(9):1750–1757.
Learn more about WELIREG: