Efficacy Data for WELIREG® (belzutifan) in Certain Adults With Clear Cell RCC

- 25% reduced risk of disease progression or death shown with WELIREG vs everolimus (HR=0.75a; 95% CI, 0.63–0.90; P=0.0008)b
- Events observed: 257/374 (69%) with WELIREG vs 262/372 (70%) with everolimus
- Median PFSc: 5.6 months (95% CI, 3.9–7.0) with WELIREG vs 5.6 months (95% CI, 4.8–5.8) with everolimus



At final analysis, OS was not statistically significant with WELIREG vs everolimus
(HR=0.92a; 95% CI, 0.77–1.10; P=NSd)
- Events observed: 254/374 (68%) with WELIREG vs 259/372 (70%) with everolimus.
- Median OS: 21 months (95% Cl, 18–24) with WELIREG vs 18 months (95% Cl, 16–22) with everolimus.
Based on the stratified Cox proportional hazard model.
One-sided P-value based on stratified log-rank test compared with the significance boundary of 0.0021.
From product-limit (Kaplan-Meier) method for censored data.
Based on stratified log-rank test.
CI = confidence interval; HR = hazard ratio; NS = not statistically significant; OS = overall survival; PD-1/L1 = programmed death receptor-1 (PD-1)/programmed death-ligand 1 (PD-L1); PFS = progression-free survival; VEGF-TKI = vascular endothelial growth factor tyrosine kinase inhibitor.

One-sided P-value based on stratified Miettinen and Nurminen (M&N) method.
ORR measured by blinded independent central review using RECIST v1.1.
ORR = objective response rate; CR = complete response; PR = partial response.




Of all evaluated patients, 49% received 2 to 3 prior VEGF receptor–targeted therapies

1L = first line; 2L = second line; 3L = third line; ECOG PS = Eastern Cooperative Oncology Group performance status; IMDC = International Metastatic Database Consortium; PD-1/L1 = programmed death receptor-1 (PD-1)/programmed death-ligand 1 (PD-L1); RCC = renal cell carcinoma; RECIST v1.1 = Response Evaluation Criteria In Solid Tumors; VEGF = vascular endothelial growth factor; VEGF-TKI = vascular endothelial growth factor tyrosine kinase inhibitor.
Extended analysis of LITESPARK-005:
PFS at 25.7-month median follow-up in adult patients with advanced ccRCC1
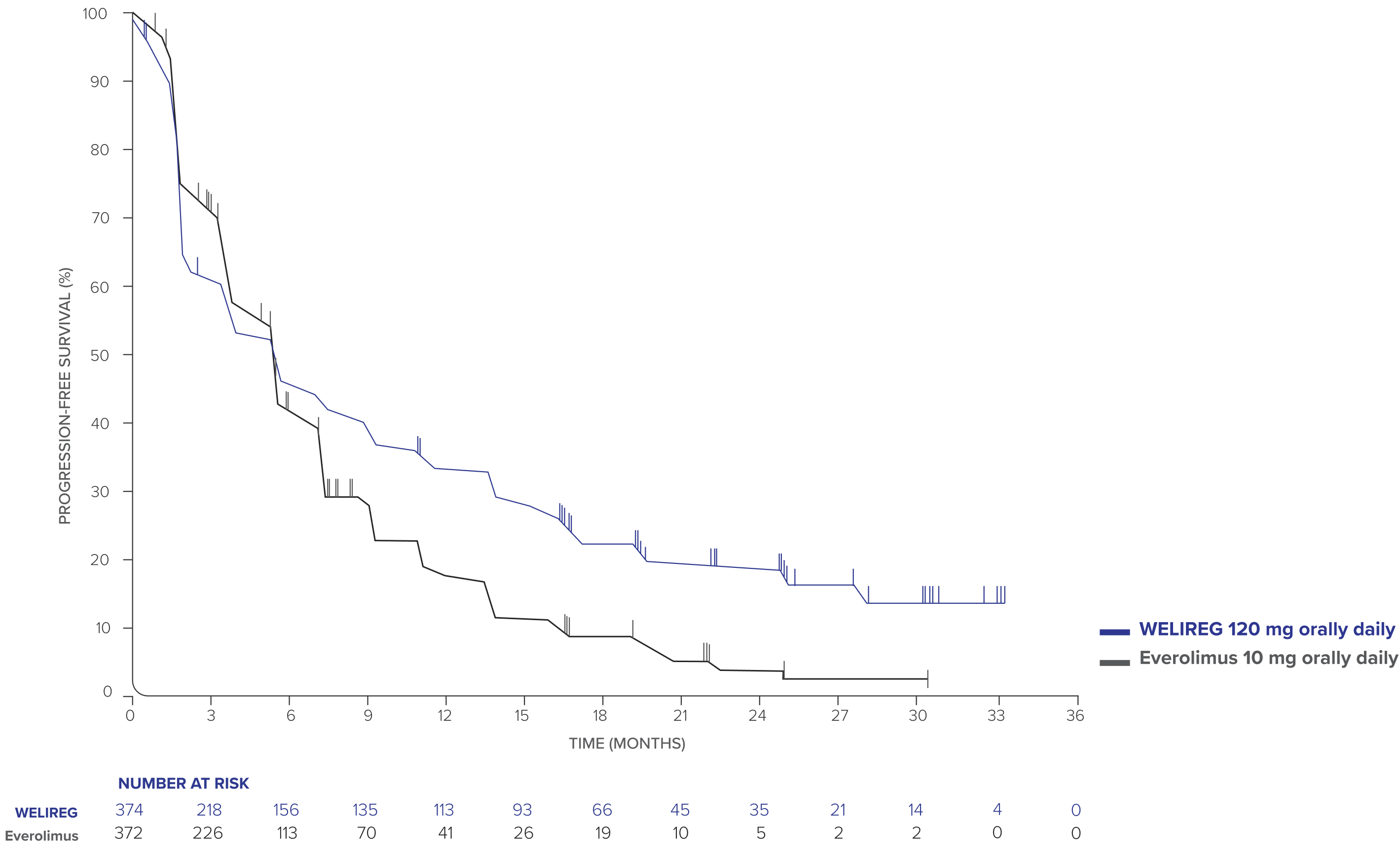

- PFS events observed: 289/374 (77.3%) with WELIREG vs 276/372 (74.2%) with everolimus.
- Median PFS: 5.6 months (95% CI, 3.8–6.5) with WELIREG vs 5.6 months (95% CI, 4.8–5.8) with everolimus.
- ORR: 22.7% (95% CI, 18.6–27.3; n=85/374) with WELIREG vs 3.5% (95% CI, 1.9–5.9; n=13/372) with everolimus.
- CR was 3.5% (n=13/374) and PR was 19.3% (n=72/374) with WELIREG; CR was 0% (n=0/372) and PR was 3.5% (n=13/372) with everolimus.
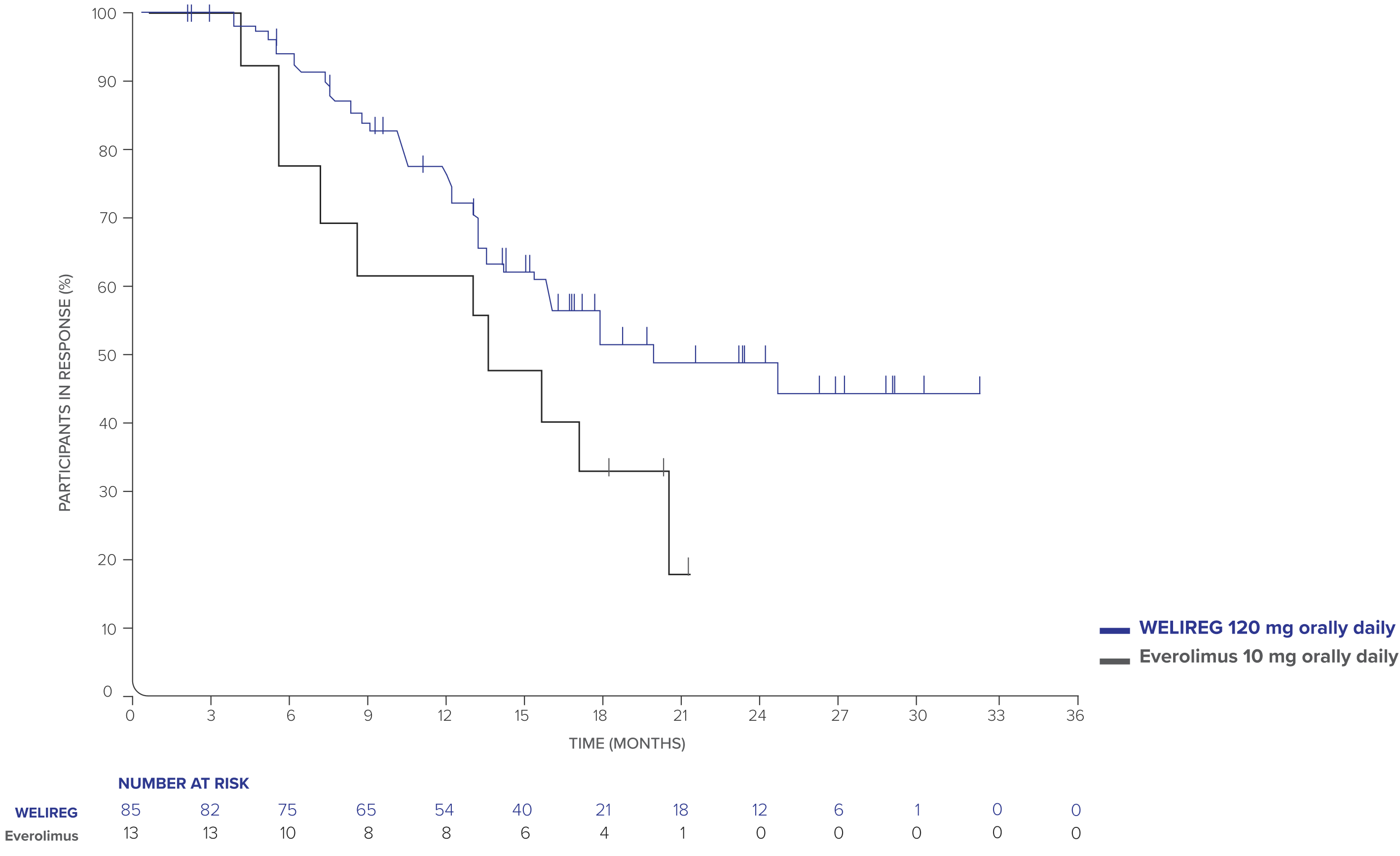

- Median DOR: 19.5 months (range: 1.9+ to 31.6+ months) with WELIREG vs 13.7 months (range: 3.8 to 21.2+ months) with everolimus.
- Median TTR: 3.8 months (range: 1.7 to 22.0 months) with WELIREG vs 3.7 months (range: 1.8 to 5.4 months) with everolimus.
TTR = time to response.
Additional resources
WELIREG may be considered as early as the second line for your appropriate adult patients with advanced ccRCC following treatment with both anti–PD-1/L1 and VEGF-TKI therapies